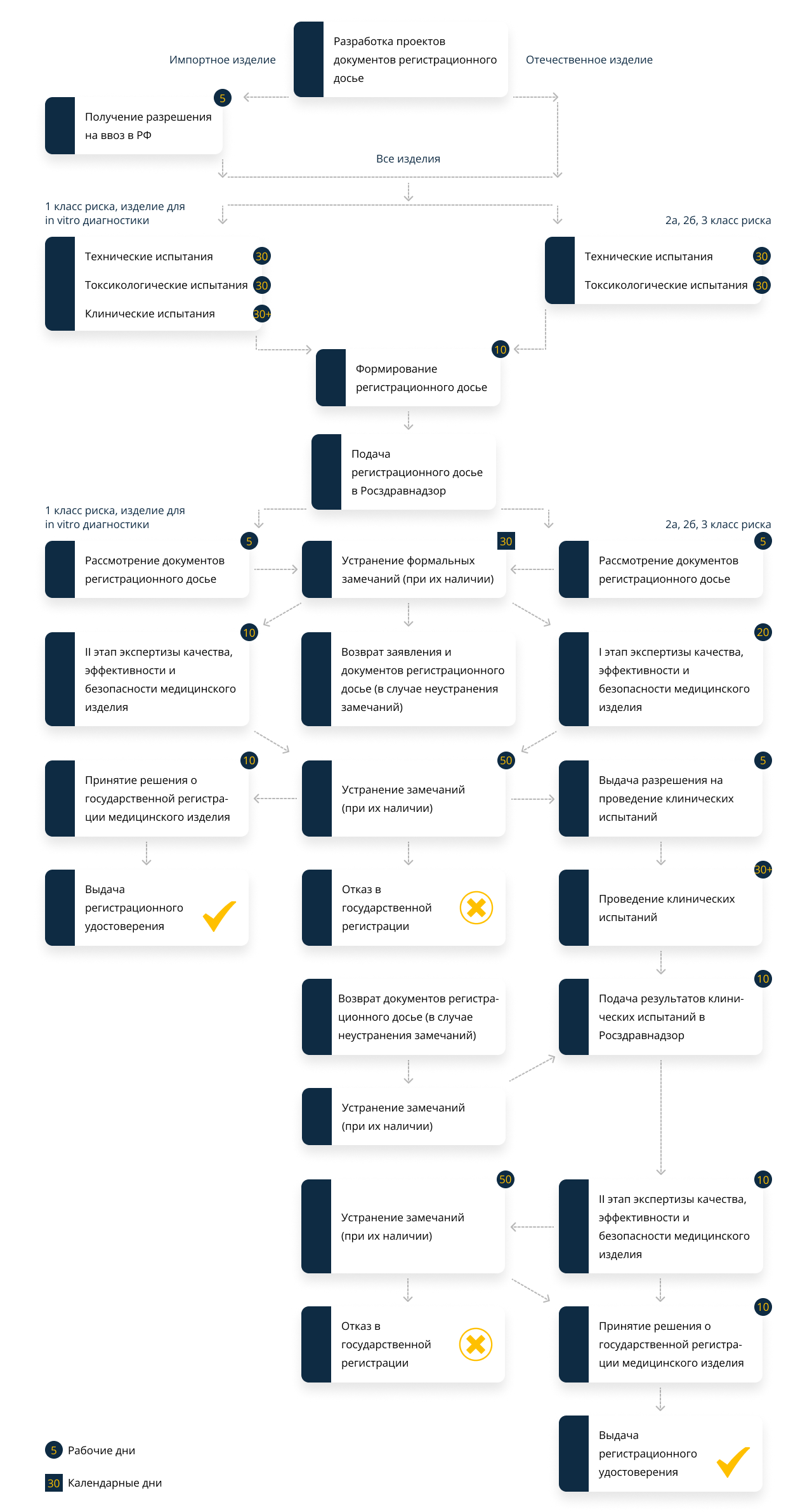
В кратчайшие сроки эксперты выполнят оценку, разработают техническую и эксплуатационную документацию, подготовят сведения о нормативной документации и заявления на регистрацию согласно требованиям, прописанным в нормативных документах. Если вы уже успели подготовить часть этих документов, специалисты оценят их соответствие прописанным нормативам, выполнят корректировку, доработку и актуализацию, если возникнет такая потребность.
Ввоз медицинских изделий иностранного производства в РФ возможен только при наличии разрешения. Эксперты подготовят комплект документации, которая необходима для получения разрешения на проведение испытаний и ввоза из-за рубежа требуемого количества образцов. Документ будет включен в состав регистрационного досье.
Мы сотрудничаем с надежными, аккредитованными лабораториями, которые проводят весь необходимый список испытаний для регистрации МИ. Чтобы ускорить регистрационные мероприятия, обеспечим одновременное выполнение технических и токсикологических исследований. Длительность клинических испытаний — 30 дней. Сроки зависят от вида мед. изделия. Если оно относится к измерительным средствам, дополнительно проводятся испытания для утверждения типа средства измерения.
Сотрудничаем с лучшими медицинскими учреждениями, на базе которых проводятся клинические испытания. Изделия, которые относятся к классу риска 2а, 2б, 3, проходят клинические испытания на основании специального разрешения на их проведение (см. в схеме ниже).
Собираются все необходимые документы, которые будут входить в состав регистрационного досье. Проверяется наличие всех подписей, печатей, виз, правильность подписания документов и полнота содержащихся в них сведений. Оплачиваются все государственные пошлины.
Комплект документов направляется в Росздравнадзор в соответствии с административной процедурой. Высший орган выполняет проверку предоставленной документации на соответствие описи.
Росздравнадзор проверяет полноту и достоверность предоставленных сведений, которые содержатся в документах досье и заявлении на регистрацию МИ
Благодаря огромной практике и опыту наших экспертов, вероятность запроса дополнительных сведений и замечаний во время проверки сведены к нулю. Но в единичных случаях Росздравнадзор может запросить дополнительные данные. В такой ситуации мы оперативно решим вопрос и обеспечим инициирование процедуры регистрации МИ.
Росздравнадзор проверяет достоверность, полноту предоставленных сведений в досье и заявлении на регистрацию МИ.
Уполномоченная организация Росздравнадзора проводит экспертизу полноты и результатов всех проведенных испытаний, в том числе и на утверждение средств измерения.
Если в течение 30 рабочих дней выявленные замечания не были устранены и (или) не предоставлены документы, Росздравнадзор делает возврат документов с обоснованием причины.
Проводится экспертная оценка заявления и рег. досье на допуск (недопуск) к проведению испытаний.
Его оформляют на основании приказа Федеральной службы по надзору в сфере здравоохранения.
От Росздравнадзора может поступить запрос на предоставление дополнительных сведений. В нашей практике подобные ситуации возникают очень редко, поскольку мы изначально при подготовке документов руководствуемся действующими нормативными документами. Если же такой вопрос поступил, эксперты оперативно подготовят ответ.
Если принимается решение выдать разрешение для испытаний, Росздравнадзор приостанавливает госрегистрацию (необходима для остановки течения сроков).
После окончания регистрационных мер выдается РУ установленного образца.
Даже если заказчики обратились после получения отказа, мы обязательно доведем процедуру регистрации до конца.
Проводятся только в учреждениях, аккредитованных на выполнение клинических испытаний мед. изделий. Список организаций и реестр выданных разрешений можно посмотреть на сайте Росздравнадзора. Мы сотрудничаем только с проверенными организациями в рамках соглашения.
Если документы представлены не в полном объеме, в них обнаружены недостоверные данные или предоставлены документы без перевода на русских язык для зарубежных МИ, возможен возврат заявления на доработку. Наши эксперты знают все тонкости в подготовке документации, поэтому вероятность возврата минимальная.
Вместе с результатами исследований направляется заявление о возобновлении госрегистрации МИ (из-за приостановки регистрации на проведение клинических исследований). Росздравнадзор проверяет полноту сведений и принимает решение о возобновлении регистрации.
В случае возврата заявления о возобновлении регистрации, устраняются все выявленные недостатки, на основании которых принималось решение о возврате.
От Росздравнадзора может поступить запрос на дополнительные сведения (в нашей практике такое случается крайне редко).
Уполномоченная организация Росздравнадзора проверяет полноту и результаты испытаний, а потом оформляет и передает в Росздравнадзор заключение по результатам качества, эффективности и безопасности мед. изделия. Успешность прохождения данного этапа регистрации гарантирована компетентностью наших экспертов и сотрудничеством с проверенными организациями, ответственными за все испытания и исследования.
Даже если заказчик получил отказ в регистрации медицинского изделия, мы доведем процедуру до конца.
Решение оформляется на основании приказа ФС по надзору в сфере здравоохранения.
После прохождения всех этапов регистрации выдается РУ.












