С 01.01.22 в России и других странах ЕАЭС начали действовать единые стандарты регистрации медизделий. Данная процедура регламентирована Решением №46 от 12.02.16. Согласно поправкам, Росздравнадзор проводит процедуру в таком порядке:
- Анализ МИ, определение класса риска и номенклатурного вида. Формирование регистрационного досье.
- Проведение лабораторных испытаний (технических, токсикологических, клинических, на электромагнитную совместимость, при необходимости) с целью подтверждения качества изделия, подготовка регистрационного досье в соответствии с правилами ЕАЭС.
- Выбор страны-референта для подачи заявления на регистрацию и стран-признания, где планируется реализация МИ.
- Внесение заявления в законодательный орган, ответственный за процесс регистрации. Проверка полученных данных осуществляется экспертами ведомства.
- Экспертиза для оценки безопасности, эффективности и качества.
- Инспектирование производства.
- Регистрация МИ. Если в результате проверки государственным органом не выявлено никаких нарушений, выдается регистрационное удостоверение. Выполняется внесение информации в Единый реестр.
?
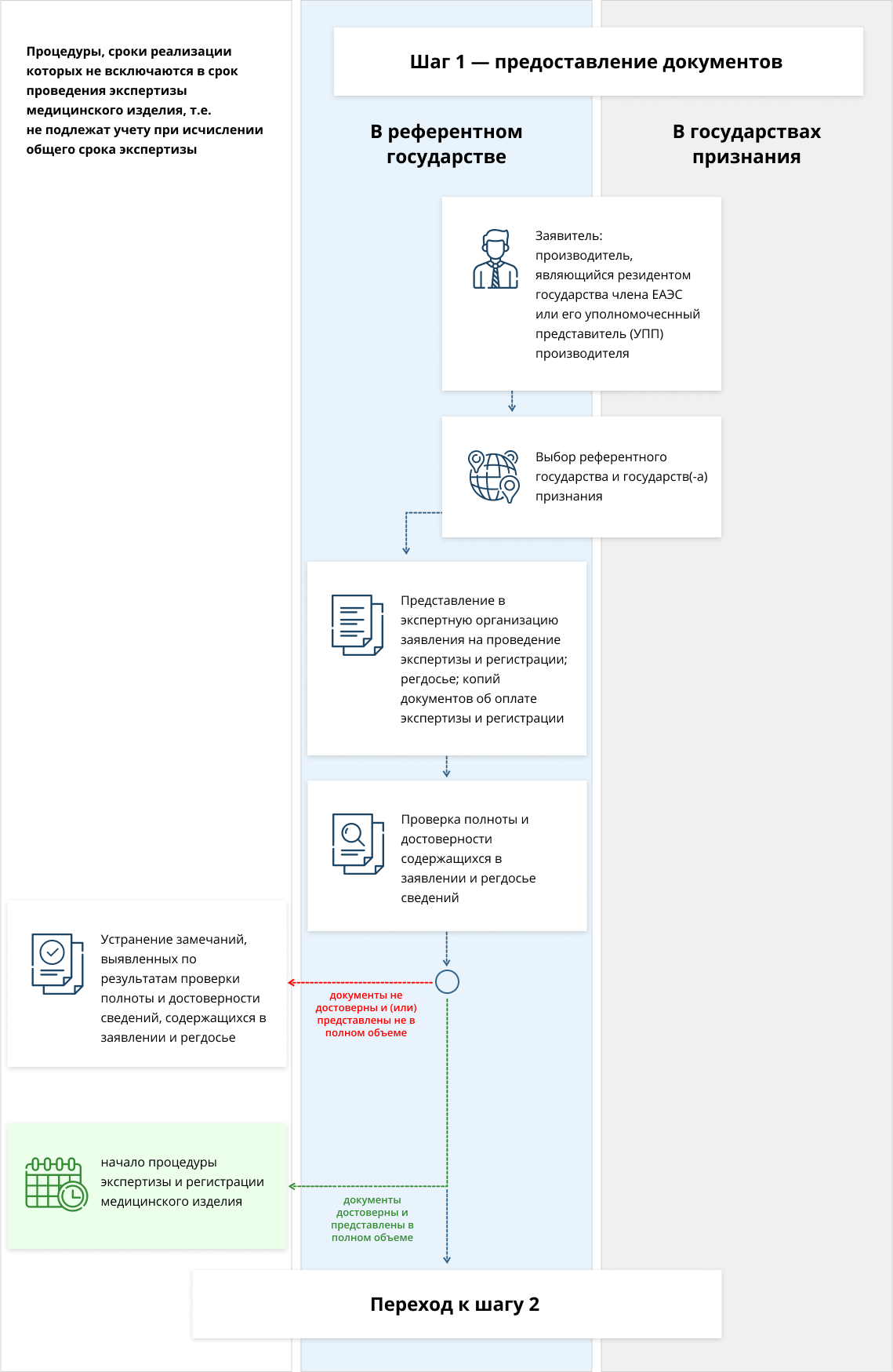
Производитель – юридическое лицо или индивидуальный предприниматель, зарегистрированный в одном из государств-участников Евроазиатского союза — Армения, Беларусь, Казахстан, Кыргызстан, Россия. Уполномоченным представителем производителя является юридическое, либо физическое лицо в статусе индивидуального предпринимателя — резидент государства-союзника, обладающее доверенностью, которая подтверждает его полномочия. Уполномоченный представитель защищает интересы производителя, отвечает за законное обращение медицинского изделия (МИ) на территории стран-участников ЕАЭС и соблюдение требований, которые предъявляются к качеству, безопасности и эффективности МИ.
?
Референтное государство – это выбранное заявителем государство-член ЕАЭС, уполномоченный орган (экспертная организация) которого будет осуществлять процедуру регистрации, а также экспертизу. В свою очередь, государство признания – это государство-член ЕАЭС, уполномоченный орган (экспертная организация) которого осуществляет процедуру согласования экспертного заключения референтного государства. Заявитель должен выбрать как минимум одно государство признания.
?
Заявитель представляет в уполномоченный орган (экспертную организацию) референтного государства регистрационное досье (РД), в состав которого входят: 1) Заявление стандартной формы о проведении экспертизы и регистрации МИ. 2) Документы, перечисленные в приложении N 4 к Правилам регистрации и экспертизы безопасности, качества и эффективности медицинских изделий. 3) Экземпляры или копии платежных документов, удостоверяющих проведение оплаты регистрационных процедур в референтном государстве.
?
Проверка полноты, достоверности и достаточности информации о МИ, содержащейся в РД, выполняется уполномоченным органом (экспертной организацией) референтного государства. На проверку отводится 7 рабочих дней, со дня подачи РД. По результатам проверки может быть принято положительное, либо отрицательное решение. В первом случае начинается процедура экспертизы и регистрации, во втором — досье возвращается на доработку для устранения обнаруженных нарушений (замечаний).
?
Срок устранения замечаний составляет 30 рабочих дней.
?
В течение 3 рабочих дней со дня представления пакета документов, а также в случае устранения замечаний в установленный срок (30 дней), уполномоченный орган (экспертная организация) референтного государства принимает решение о начале процедур регистрации и экспертизы.
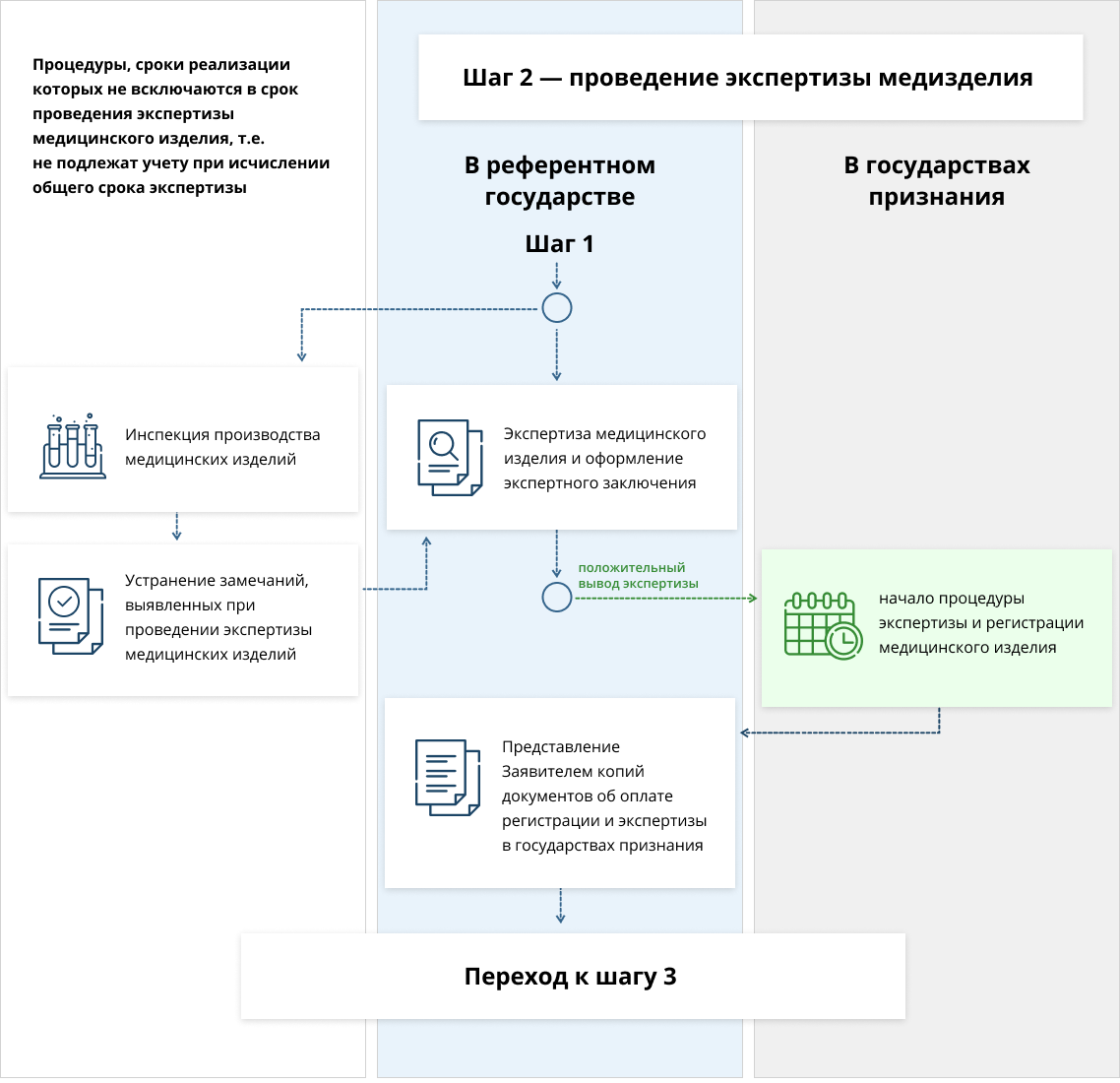
После первого шага документы передаются на экспертизу. Одновременно с этой процедурой уполномоченный законодательством орган проводит проверку производства МИ.
?
Экспертиза занимает 45 рабочих дней с момента принятия решения уполномоченного органа о начале процедур регистрации и экспертизы.
?
Для подготовки экспертного заключения уполномоченный орган или назначенная им организация инспектирует условия производства регистрируемого МИ. Срок проведения инспектирования производства не должен в совокупности превышать 90 рабочих дней со дня принятия решения о начале инспектирования производства.
?
В случае выявления замечаний к содержанию заявления и (или) составу РД, уполномоченный орган (экспертная организация) направляет заявителю запрос, в котором указывает все обнаруженные нарушения и возможные способы их ликвидации. На устранение выявленных недостатков дается 30 дней. При расчете сроков экспертизы не учитывается временной интервал с момента отправки запроса до момента устранения замечаний.
?
При положительных результатах экспертизы в течение 5 рабочих дней уполномоченный орган (экспертная организация) направляет заявителю уведомление о необходимости представления копий документов об оплате процедуры согласования экспертного заключения в государствах признания. На выполнение этого действия законом отведено 30 календарных дней.
?
Без предоставления копий платежных документов экспертные и регистрационные мероприятия в государствах-союзниках не начинаются.
 Проверка выполняется до момента формирования заключения экспертной оценки!
Проверка выполняется до момента формирования заключения экспертной оценки!
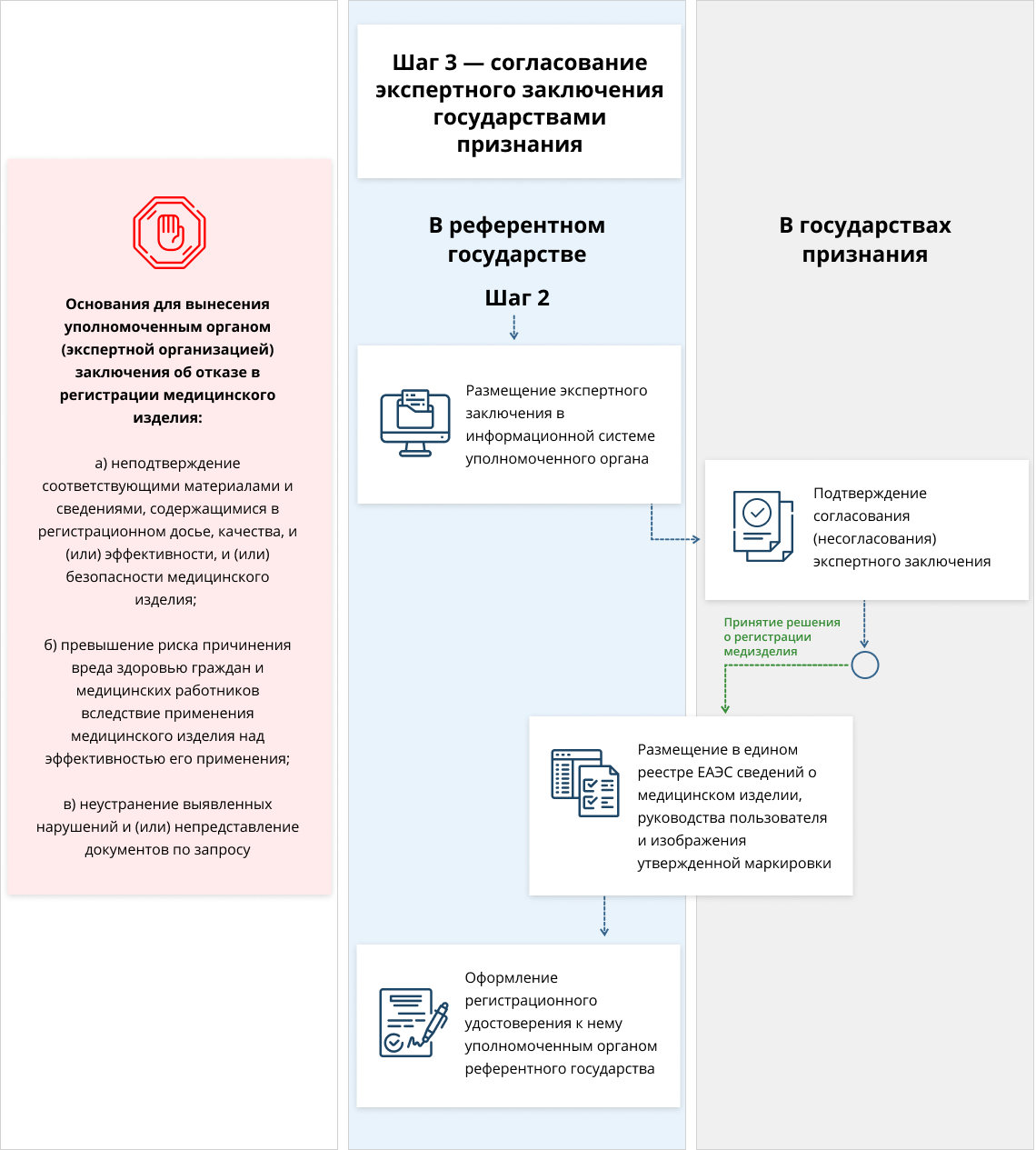
Третий шаг — согласование заключения экспертов с государствами-членами.
?
Референтное государство публикует экспертное заключение в информационной системе, которая доступна для уполномоченных органов (экспертных организаций) государств признания.
?
Уполномоченный орган (экспертная организация) государства (или государств) признания рассматривает экспертное заключение в течение 30 рабочих дней, а затем направляет в уполномоченный орган (экспертную организацию) референтного государства подтверждение согласования экспертного заключения. В случае непредставления государством признания подтверждения согласования (несогласования) экспертного заключения в течение одного месяца, экспертное заключение считается согласованным при условии, что заявителем были соблюдены все требования действующего законодательства.
?
При отсутствии или урегулировании разногласий между референтным государством и государствами признания по вопросу согласования экспертного заключения уполномоченный орган (экспертная организация) референтного государства принимает решение о регистрации медицинского изделия и размещает соответствующую информацию в едином реестре медицинских изделий, зарегистрированных в рамках Союза
?
В течение 10 рабочих дней со дня принятия решения о регистрации медицинского изделия уполномоченный орган (экспертная организация) референтного государства оформляет и выдает заявителю РУ.
Если результат согласования будет положительным, оформляется РУ и приложения к нему.
Сроки лицензирования по ЕАЭС
Продолжительность регистрации МИ по правилам ЕЭК ЕАЭС составляет от 12 до 24 месяцев. Сроки зависят от класса и степени риска изделия, количества вариантов исполнения и иных особенностей МИ. В отличие от регистрации, осуществляемой по национальным правилам, процедура по правилам ЕАЭС подразумевает более сложный и дорогой процесс, по этой причине регистрация занимает больше времени.
|
ЕАЭС |
РФ |
| Срок действия РУ |
Бессрочное.
|
Действует до 2026 года
|
| Территория признания РУ |
Во всех странах признания. Их выбирает заявитель. Минимум одна. Например, Россия +
Казахстан (страна-признания).
|
В России
|
| Инспекция производства |
1 и 2а (не стерильные) - добровольная, 2а (стерильные), 2б и 3 - обязательная.
|
Нет
|
| Согласование экспертного
заключения |
Со странами признания (Армения, Белоруссия, Казахстан, Киргизия, Россия).
|
Нет
|
| Госпошлины |
В РФ и странах признания пошлина будет оплачиваться в стране регистрации и в каждой
стране признания, которые укажет заявитель.
|
В РФ
|
Что получает заявитель при регистрации
Сотрудничая с нами, заявитель сможет сэкономить не только средства, но и время. Вы получите официально оформленное регистрационное удостоверение, согласно которому можно вводить в оборот МИ в странах-участниках ЕАЭС. С нами ваша продукция станет конкурентной, что позволит получить максимальную выгоду от ее продаж.
Оставьте заявку на услугу
Мы свяжемся с вами в ближайшее время и ответим на все интересующие вопросы.
Заказать услугу
Особенности перехода с национальных правил регистрации
Согласно Решению Совета ЕЭК No46 от 12.02.2019 г. и по распоряжению Коллегии ЕЭК от 9 марта 2021 г. N 28 "О проекте распоряжения Совета Евразийской экономической комиссии «О проекте Протокола о внесении изменения в Соглашение о единых принципах и правилах обращения медицинских изделий в рамках Евразийского экономического союза от 23 декабря 2014 года», до 31.12.2021 г. заявление об экспертизе или регистрации можно было подавать по законодательной форме страны. Сроки годности удостоверений зависят от страны, где изделие регистрировалось.
Удостоверения, полученные по правилам стран-участников после 31.12.2021 г., продолжат действовать до конца их срока годности:
Армения
процедура не действует, не регламентирована
МИ, которые прошли регистрацию по правилам системы законодательства партнера (исключением являются оформленные бессрочно и имеющие подтверждение) подлежат перерегистрации по требованиям совета ЕАЭС. В таком случае повторная процедура проводится точно по таким же правилам, как и первая регистрация. Наличие действующего РУ немного упрощает процедуру доказательства качества, безопасности и эффективности.
Можно вносить новые данные или изменения в имеющуюся документацию, зарегистрированную по действующим законам принимающей страны. Для начала процедуры заявления принимаются до 21.12.26 г.

















